
Indice
La fibrosi cistica, indicata anche con la sigla FC, è una malattia cronica grave che interessa l’apparato respiratorio e quello digerente.
In questo articolo ne analizzeremo le cause, i sintomi e prenderemo in esame i trattamenti disponibili.
Cos'è la fibrosi cistica?
La fibrosi cistica è una malattia genetica ereditaria che colpisce principalmente i polmoni, ma può interessare anche il pancreas, il fegato, l’intestino e altre parti del corpo. Si tratta di una condizione cronica e progressiva che può avere gravi conseguenze sulla salute.
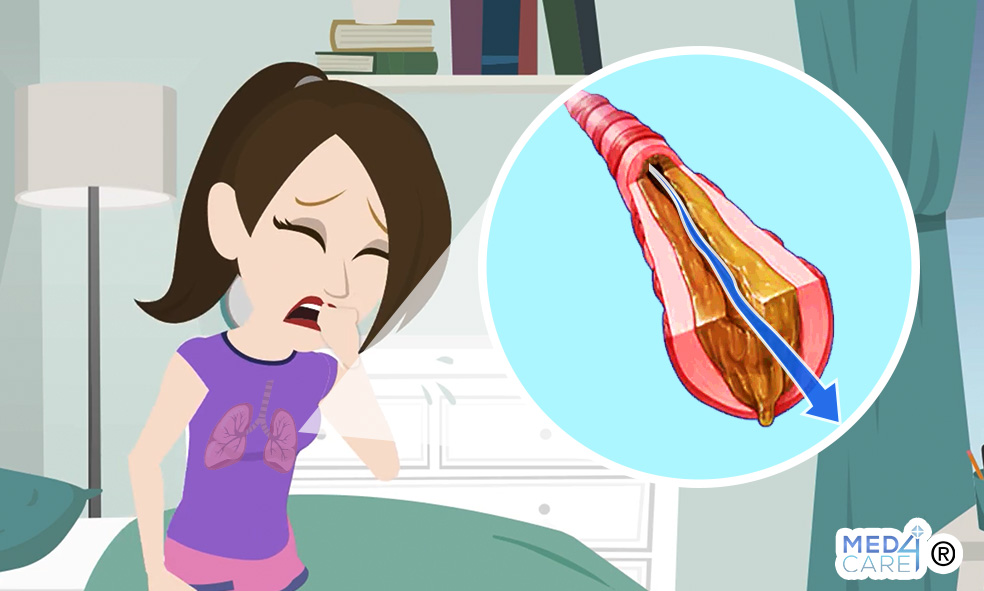
All’origine di questa patologia c’è un alterato valore di acqua e sale nelle membrane cellulari. Ciò porta alla produzione di un muco viscoso e denso nelle vie respiratorie e nell’apparato digerente.
Nei polmoni queste secrezioni possono non solo rendere difficile la respirazione, ma anche intrappolare le particelle di polvere e i batteri causando infezioni ricorrenti.
Nel pancreas il muco tende a intasare i dotti pancreatici, impedendo l’uscita delle sostanze digestive nel tratto intestinale e causando problemi di digestione e malnutrizione.

Quanto è frequente la fibrosi cistica?
Sebbene sia classificata come una malattia rara, la fibrosi cistica è molto diffusa e colpisce all’incirca un neonato ogni 2500-3000 nel mondo.
La sua incidenza varia a seconda della regione geografica e dell’etnia di appartenenza ma, consultando i dati statistici, risulta più comune tra le persone di etnia caucasica e meno diffusa tra quelle di etnia africana, asiatica e ispanica.
Cause della fibrosi cistica
La principale causa della fibrosi cistica è una mutazione del gene CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) che ha il compito di regolare il flusso di sale e acqua nelle membrane cellulari. Nelle persone affette da fibrosi cistica questo gene non funziona correttamente e ne consegue la secrezione di muco denso e viscoso, che compromette il sistema respiratorio e digestivo.
La fibrosi cistica è ereditaria e viene trasmessa da genitori portatori di una mutazione del gene CFTR. Se entrambi i genitori sono portatori della mutazione, ogni figlio ha una probabilità del 25% di ereditare la malattia. Se, invece, solo uno dei due è portatore della mutazione, ogni figlio ha una probabilità del 50% di ereditare a sua volta la mutazione del gene.
Esistono più di 2000 mutazioni del gene CFTR, le quali possono presentarsi in varie combinazioni: la gravità della patologia dipende proprio dalle diverse combinazioni possibili.
Sintomi della fibrosi cistica
I sintomi attraverso i quali può manifestarsi la fibrosi cistica sono numerosi e variano da persona a persona. Compaiono progressivamente e tendono a peggiorare con l’avanzamento dell’età.
Quelli che coinvolgono l’apparato respiratorio includono:
- tosse cronica;
- respiro sibilante;
- difficoltà respiratorie, specialmente durante l’esercizio fisico;
- infezioni polmonari ricorrenti, come la bronchite e la polmonite;
- produzione di espettorato denso e viscoso.
I sintomi dell’apparato digerente comprendono:
- problemi digestivi, come la diarrea, la stitichezza, il gonfiore addominale e la flatulenza;
- malnutrizione e perdita di peso;
- insufficienza pancreatica, che può causare problemi nella digestione dei grassi e delle proteine.
Possono manifestarsi anche altri sintomi associati, tra cui:
- sudorazione eccessiva e salata;
- infertilità maschile e femminile;
- disturbi ossei, come l’osteoporosi;
- problemi epatici, come l’ittero e l’insufficienza epatica.
Diagnosi di fibrosi cistica
Nella maggior parte dei casi la fibrosi cistica viene diagnosticata alla nascita grazie a uno screening neonatale, che prevede un test della tripsina (un enzima proteolitico) e un’analisi della sudorazione, che misura la concentrazione di sale nel sudore del neonato.
Tuttavia vi sono casi in cui i sintomi della malattia possono insorgere durante o dopo l’età infantile. Pertanto la diagnosi verrà effettuata da un medico specializzato in malattie polmonari, da uno specialista in genetica medica o da un pediatra.
Anamnesi
L’anamnesi è un’importante valutazione medica, che consiste in un colloquio riguardante la salute generale del paziente e i sintomi della malattia.
Durante questa fase il medico non solo raccoglie informazioni sulle condizioni mediche passate e presenti del paziente, ma verifica anche l’eventuale presenza di malattie genetiche nella sua famiglia.
Gli aspetti che verranno presi maggiormente in considerazione riguardano:
- la presenza nota di una mutazione del gene CFTR da parte di uno o entrambi i genitori;
- eventuali problemi respiratori o digestivi cronici;
- insufficienza pancreatica;
- la bassa statura o la scarsa crescita durante l’infanzia.
Qualora il colloquio anamnestico dovesse destare il sospetto di fibrosi cistica, il medico può raccomandare un test del sudore o un esame genetico, in modo da confermare la diagnosi.
Esame obiettivo
L’esame obiettivo è essenziale per identificare tempestivamente la presenza della fibrosi cistica e avviare il trattamento necessario a prevenire eventuali complicanze. Durante il controllo, il medico valuta le funzioni respiratorie e digestive del paziente e controlla la presenza di infezioni.
Nello specifico vengono impiegate le seguenti procedure:
- Auscultazione polmonare: il medico ascolta il respiro del paziente con un fonendoscopio per valutare eventuali rumori anomali, come respiro sibilante o crepitii polmonari.
- Esame della cute: viene valutata la presenza di sudorazione eccessiva e salata, tipica di chi è affetto da fibrosi cistica.
- Esame addominale: lo specialista palpa l’addome per verificare la presenza di eventuali masse, di gonfiore addominale oppure di organi ingrossati.
- Misurazione dell’indice di massa corporea (BMI): attraverso la misurazione dell’altezza e del peso del paziente viene calcolato l’indice di massa corporea, un indicatore della salute generale e della nutrizione.
Esame genetico
L’esame genetico è uno strumento preciso ed efficace che ha l’obiettivo di identificare le mutazioni del gene CFTR. È necessario per confermare la diagnosi di fibrosi cistica, soprattutto quando il test del sudore ha dato esito incerto o negativo, ma si manifestano comunque i sintomi tipici della malattia.
Il test può essere effettuato su campioni di sangue, saliva o tessuti prelevati tramite biopsia.
Questo esame può non soltanto rilevare la presenza della malattia in un soggetto, ma anche quantificare il rischio di avere un figlio affetto da fibrosi cistica o di essere portatore della mutazione genetica.
Esami strumentali per la fibrosi cistica
Si tratta di tutti quegli esami necessari a monitorare la funzione respiratoria, valutare eventuali complicanze e controllare l’efficacia del trattamento.
Gli esami strumentali più comuni per la fibrosi cistica sono:
- Spirometria: un esame non invasivo che serve a svolgere misurazioni di volumi e flussi polmonari; il paziente dovrà inspirare profondamente e poi espirare con forza in un boccaglio collegato allo spirometro.
- Tomografia computerizzata (TC) del torace: la TC è un esame di imaging medico che utilizza raggi X per creare immagini dettagliate dei polmoni e dell’apparato respiratorio.
- Broncoscopia: prevede l’inserimento di un tubo sottile e flessibile attraverso la bocca o il naso per visualizzare l’interno delle vie respiratorie.
- Ecografia addominale: utilizza onde sonore ad alta frequenza per creare immagini di organi interni come il fegato, il pancreas e i reni.
Diagnosi differenziale della fibrosi cistica
La diagnosi differenziale è un processo medico che aiuta a escludere altre malattie che si manifestano con sintomi simili a quelli della fibrosi cistica. Si tratta di patologie che coinvolgono il sistema respiratorio e digestivo a diversi livelli, ma che non comportano la produzione di muco denso.
Queste patologie possono essere:
- Asma: si tratta di una malattia respiratoria che può causare respiro sibilante, tosse e difficoltà respiratorie.
- Cigliopatie o Ciliopatie: sono malattie genetiche rare che causano complicazioni respiratorie simili alla fibrosi cistica. La causa primaria di queste malattie risiede nella struttura delle ciglia, non nella produzione di muco.
- Fibrosi polmonare idiopatica: questa patologia provoca cicatrici sui polmoni e la conseguente formazione di tessuto cicatriziale, portando alla comparsa di difficoltà respiratorie.
- Disordini dell’immunità: si tratta di malattie in cui il sistema immunitario attacca erroneamente i tessuti sani del corpo generando problemi respiratori e digestivi.
Trattamento della fibrosi cistica
Nonostante si tratti di una patologia cronica, molti pazienti affetti da fibrosi cistica hanno la possibilità di vivere meglio grazie ai progressi raggiunti nella gestione dei sintomi e nella cura delle infezioni polmonari.
I trattamenti prevedono un approccio multidisciplinare che coinvolge terapie farmacologiche, respiratorie e digestive.
Terapie farmacologiche
Questo tipo di cure prevede l’assunzione di:
- Antibiotici: necessari a trattare le infezioni respiratorie; vengono somministrati per via inalatoria o per via orale e possono essere utilizzati in cicli o in modo continuo.
- Mucolitici: aiutano a ridurre la viscosità dell’espettorato nei polmoni; possono essere somministrati per via inalatoria o per via orale.
- Broncodilatatori: dilatano le vie respiratorie così da facilitare la respirazione.
- Antinfiammatori: agiscono sui polmoni per ridurne l’infiammazione.
Terapie respiratorie
Le terapie respiratorie sono finalizzate a migliorare la funzione polmonare, a prevenire le infezioni e ad alleviare i sintomi che si manifestano a livello respiratorio.
Le principali sono:
- Fisioterapia respiratoria: consiste in un insieme di esercizi che aiutano il paziente a espellere il muco dai polmoni, migliorando la funzione polmonare e prevenendo l’insorgere di eventuali infezioni.
- Ventilazione meccanica: prevede l’utilizzo di una macchina in grado di assistere il paziente nella respirazione.
- Ossigenoterapia: viene utilizzata per fornire ossigeno supplementare ai pazienti che hanno difficoltà a respirare a sufficienza.
Terapie digestive
Le terapie digestive prevedono la somministrazione di enzimi pancreatici e integratori vitaminici al fine di migliorare la digestione e l’assorbimento dei nutrienti del paziente.
Vediamo nel dettaglio il loro funzionamento:
- Enzimi pancreatici: la loro assunzione è fondamentale dal momento che la fibrosi cistica causa un malfunzionamento della produzione di enzimi digestivi da parte del pancreas, rendendo difficile per il corpo digerire e assorbire i nutrienti.
- Integratori vitaminici: sono necessari in quanto la fibrosi cistica può determinare un malassorbimento di vitamine liposolubili, come la A, la D, la E e la K.
Queste sostanze devono costituire un’integrazione a una dieta varia e ricca di calorie e proteine, fondamentale per contrastare la perdita di peso e la malnutrizione causate dalla fibrosi cistica.
Terapie chirurgiche
Le terapie chirurgiche possono rivelarsi inevitabili in caso di complicanze gravi sia a livello respiratorio che digestivo.
Il trapianto di polmone è un’opzione necessaria per quei pazienti nei quali il funzionamento polmonare risulta irrimediabilmente compromesso.
L’intervento chirurgico a livello dell’apparato digestivo è invece previsto in caso di grave ostruzione intestinale, dovuta a un accumulo di secrezioni dall’intestino.
Si tratta chiaramente di procedure a cui si ricorre in casi estremi e che comunque non possono curare la fibrosi cistica. Inoltre bisogna tenere presenti i possibili effetti collaterali legati a un intervento chirurgico (come sanguinamento, infezione e dolore nella zona dell’operazione) e considerare anche il rischio di rigetto nel caso del trapianto.
Tuttavia entrambi gli interventi possono giovare significativamente, soprattutto il trapianto polmonare, che andrà a migliorare in modo considerevole la capacità respiratoria del paziente.
Terapie psicologiche e sociali
La terapia psicologica è importante per supportare non solo la persona affetta da fibrosi cistica, ma anche la sua famiglia. L’obiettivo principale è imparare a gestire gli aspetti emotivi e sociali della malattia, come lo stress e l’ansia.
Gli interventi psicologici possono essere di diverso tipo, tra cui:
- Terapia cognitivo comportamentale: aiuta le persone con fibrosi cistica ad adottare comportamenti positivi per gestire la malattia.
- Terapia di sostegno: fornisce un supporto emotivo ai pazienti, alle loro famiglie e può aiutare a gestire le difficoltà emotive associate alla malattia.
- La terapia familiare: aiuta a migliorare la comunicazione e la collaborazione tra i familiari del paziente.
- Terapie sociali: aiutano le persone con fibrosi cistica a mantenere una vita attiva attraverso l’inserimento in attività sociali e l’accesso a risorse, come i servizi di assistenza domiciliare e i programmi di supporto tra pari.
Fibrosi cistica: punti chiave
La fibrosi cistica è una malattia genetica che colpisce le vie respiratorie e digestive. È causata da una mutazione nel gene CFTR, che regola la produzione di muco e l’equilibrio del sale nel corpo. Può causare problemi respiratori, digestivi e nutrizionali e può portare a complicanze a lungo termine.
La diagnosi della fibrosi cistica coinvolge una serie di test diagnostici, tra cui il test del sudore e l’esame genetico. Il trattamento della malattia è personalizzato e coinvolge una combinazione di terapie e altre forme di supporto medico.
Non esiste una cura per la fibrosi cistica, ma i trattamenti attuali possono aiutare a ottimizzare le funzioni polmonari e digestive, prevenire le infezioni e migliorare la qualità della vita dei pazienti.