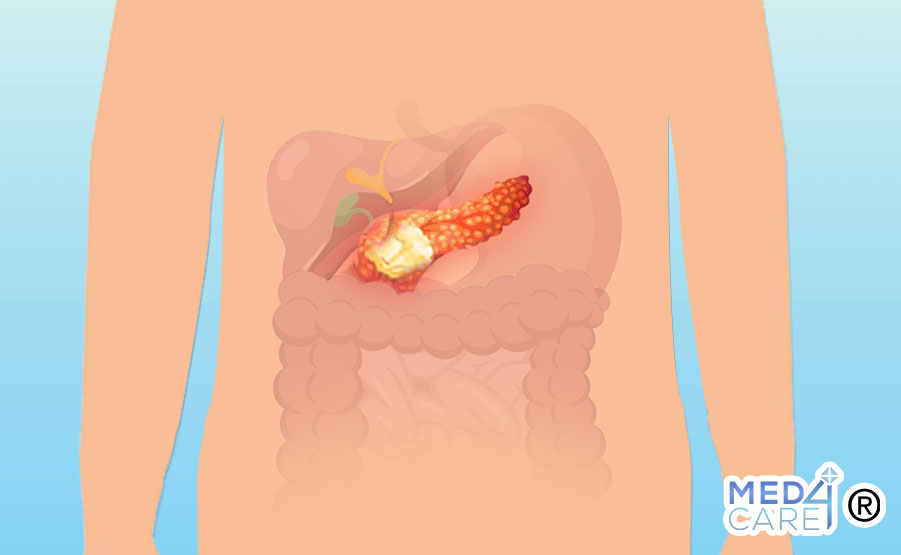

I TUMORI NEUROENDOCRINI DEL PANCREAS (PNET)
Che cosa sono i tumori neuroendocrini del pancreas
I tumori neuroendocrini del pancreas (PNET) sono delle neoplasie che bersagliano il cosiddetto pancreas endocrino, ossia quella porzione del pancreas che ha la funzione di produrre e mettere in circolo vari ormoni.
I tumori neuroendocrini del pancreas sono tra le neoplasie neuroendocrine più comuni ma rappresentano, al contempo, alcuni dei più rari tumori che possono colpire direttamente il pancreas, significativamente meno frequenti dell’adenocarcinoma pancreatico.
I tumori neuroendocrini del pancreas sono così denominati in quanto vanno a interessare le cellule neuroendocrine pancreatiche: esse sono un tipo di cellule facenti parte sia del sistema nervoso sia del sistema endocrino, perchè hanno la capacità di sintetizzare ormoni.
Il pancreas: localizzazione e funzioni
Il pancreas è una ghiandola della lunghezza compresa tra i 13 e i 15 centimetri, di colore rosato, che si localizza in una posizione difficilmente raggiungibile chirurgicamente, ben nascosta da altri visceri intestinali. Questa ghiandola è infatti collocata in posizione retroperitoneale e posteriormente si proietta tra le prime due vertebre lombari.
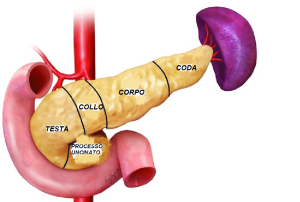
Il pancreas si suddivide anatomicamente in:
- Testa, accolta nella porzione concava del primo segmento intestinale, ossia nel duodeno;
- Collo, rivestito dal peritoneo e in rapporto con la vena porta;
- Corpo, rivestito quasi per intero dal peritoneo e situato davanti al rene sinistro e alla vena lienale;
- Coda, che si localizza davanti al rene sinistro e risale fino all’altezza della settima costa;
- Processo uncinato, che si porta davanti all’aorta addominale.
Il pancreas viene definito una ghiandola “anficrina”, dal momento che è sia implicato nei processi endocrini che nei processi esocrini.

La funzione esocrina del pancreas
Nella sua funzione esocrina, essendo una ghiandola annessa al sistema gastro-intestinale, il pancreas elabora una grande quantità di liquido ogni giorno, definito succo pancreatico. Il liquido pancreatico è ricco in enzimi che provvedono a digerire le macromolecole ingerite con il cibo, e viene riversato a livello del duodeno.
La funzione endocrina del pancreas
Nella sua funzione endocrina il pancreas provvede alla sintesi e al riversamento in circolo di alcuni ormoni, tramite l’intervento di gruppi di cellule specializzate, che prendono il nome di Isolotti del Langerhans. Queste cellule producono diversi ormoni importanti per l’organismo, come l’insulina, il glucagone, la somatostatina e il polipeptide pancreatico. [1]
Cenni di epidemiologia dei PNET
I tumori neuroendocrini del pancreas non presentano differenze, nella loro incidenza a livello globale, per quanto riguarda l’area geografica o il sesso; i PNET esibiscono, allo stesso tempo, un comportamento molto variabile, potendo sviluppare un fenotipo benigno o un fenotipo maligno. In genere, però, la grande parte dei PNET diagnosticati si evolve verso un comportamento tendenzialmente maligno.
Negli ultimi venti anni il tasso di incidenza di questo tipo di tumori è grossomodo raddoppiato probabilmente per via della nostra maggiore capacità di diagnosticarli. Ciò è dovuto per via dell’evoluzione tecnologica degli strumenti per la diagnosi e all’aumento delle conoscenze nella materia. Attualmente alcune stime indicano che la prevalenza dei tumori neuroendocrini del pancreas si attesta tra 1 e 25 casi ogni 100.000, con un’età media alla diagnosi compresa tra i 55 e i 60 anni.
Di tutti i PNET, il 10-30 % sono considerati PNET funzionali, ossia tumori nei quali si verificano sindromi ormonali, dovute all’iper-secrezione degli ormoni prodotti dalle cellule tumorali. Al contrario, nei PNET non funzionali, nei quali non si verificano sindromi ormonali, la diagnosi è più difficoltosa, venendo diagnosticati per caso dopo i 60 anni di età. [2],[3]
Eziologia dei tumori neuroendocrini del pancreas
Il completo meccanismo patogenetico attraverso cui i tumori neuroendocrini del pancreas si originano non è ancora stato del tutto acclarato, sebbene le ricerche in questo ambito stiano aumentando negli ultimi anni.
Circa il 10 % di tutti i PNET si riscontrano in associazione a varie sindromi tumorali endocrine familiari; la caratteristica di queste sindromi tumorali è la perdita di un gene oncosoppressore, vale a dire di un gene che inibisce la crescita cellulare e la replicazione, come MEN1. Venendo dunque a mancare il freno alla crescita cellulare, le cellule sono più libere di potersi replicare, aumentando la massa del tumore.
Le sindromi tumorali endocrine familiari più comunemente associate al contemporaneo riscontro di PNET sono:
- Neoplasia endocrina multipla di tipo 1 (MEN1);
- Malattia di Von Hippel-Lindau (VHL);
- Neurofibromatosi familiare di tipo 1 (NF-1);
- Sclerosi tuberosa (TSC).
A parte la familiarità per tumori neuroendocrini o per sindromi tumorali endocrine, la maggior parte dei PNET si instaura a causa di mutazioni e alterazioni geniche sporadiche in geni oncosoppressori o in proto-oncogeni.
Come nel caso di MEN1, il gene oncosoppressore è un gene che inibisce e blocca la proliferazione cellulare, quindi quando questo gene non è più funzionale (a causa di mutazioni), perde la propria capacità “frenante” sulla replicazione.
Al contrario, il proto-oncogene è un gene che facilita i processi di crescita e di differenziazione cellulare: quando una mutazione lo modifica in senso neoplastico, il gene diviene onco-gene e invia segnali in eccesso per indurre e sostenere la crescita cellulare. [2]
Tumori neuroendocrini del pancreas di tipo funzionale e non funzionale
I tumori neuroendocrini del pancreas possono innanzitutto classificarsi in:
- Tumori neuroendocrini del pancreas funzionali;
- Tumori neuroendocrini del pancreas non funzionali.
La differenza tra i due tipi di tumore risiede nella capacità di manifestare la sindrome da iper-secrezione ormonale a livello clinico.
Ad esempio quando le cellule che producono insulina (le cellule ß) acquisiscono un comportamento neoplastico tendono a produrre un eccesso di insulina, che verrà riversata nel sangue. Se l’insulina prodotta in esubero determina degli effetti sistemici negativi nell’organismo, allora si parla di tumore neuroendocrino del pancreas (insulinoma) di tipo funzionale.
Quando, invece, la quantità di ormoni prodotta dal tumore non determina una sindrome da iper-secrezione ormonale si parlerà di tumore neuroendocrino del pancreas di tipo non funzionale.
Che differenze ci sono tra i due tipi di tumore neuroendocrino?
I PNET funzionali, determinando dei sintomi sistemici causati dall’eccesso di ormoni in circolo, è più semplice che vengano diagnosticati in maniera più tempestiva, impostando in tempi rapidi la giusta terapia.
Al contrario, i PNET non funzionali, che rappresentano la maggioranza, possono rimanere silenti per lungo tempo o essere scoperti tardivamente per via di altri segni e sintomi più aspecifici, come ittero ostruttivo e altri sintomi riconducibili a una pancreatite. Molto spesso i PNET non funzionali vengono diagnosticati in pazienti asintomatici in maniera del tutto fortuita, in seguito a indagini per altri scopi. [2], [3]
Classificazione biologica dei tumori neuroendocrini del pancreas
I tumori neuroendocrini del pancreas possono classificarsi a seconda del tipo di cellula ad attività endocrina interessata dalla neoplasia, e dunque dal tipo di ormone da essa elaborato.
La classificazione biologica (o funzionale) dei PNET è stata sviluppata dall’Organizzazione Mondiale della Sanità (OMS) nel 2010 ed è riassunta nella tabella riepilogativa riportata di seguito:
| Denominazione | Tipo di cellula | Ormoni secreti | Sintomi correlati | Potenziale di malignità |
| Insulinoma | ß (o B) | Insulina | Ipoglicemia | 10 % |
| Gastrinoma | G | Gastrina | Ulcera peptica severa | 60 % – 90 % |
| VIPoma | D1 | Peptide intestinale vasoattivo | Diarrea acquosa, ipokaliemia, acidosi | 40 % – 70 % |
| Glucagonoma | ɑ (o A) | Glucagone | Intolleranza al glucosio, stomatite | 50 % – 80 % |
| Somatostatinoma | δ (o D) | Somatostatina | Iperglicemia, steatorrea | 70 % |
Dei vari tipi di tumori neuroendocrini pancreatici riportati, l’insulinoma rappresenta sicuramente la tipologia di più frequente riscontro, seguito in ordine decrescente dai gastrinomi, dai glucagonomi, dai VIPomi e dai somatostatinomi; sono poi presenti ulteriori tumori molto più rari, come il PPoma.
In generale, come si evince, tutti i tumori neuroendocrini del pancreas hanno una grande probabilità di trasformarsi in neoplasie maligne, ad eccezione dell’insulinoma che di norma è benigno. [2], [3]
Sintomi dei tumori neuroendocrini del pancreas di tipo funzionale
I sintomi provocati dai tumori neuroendocrini del pancreas di tipo funzionale, ovvero da quei tumori che inducono una sindrome da iper-secrezione di ormoni, vengono a dipendere dal tipo di ormone secreto dalle cellule neoplastiche. Infatti, a seconda dell’ormone secreto (in eccesso) e delle sue attività biologiche, nonché in base all’interazione con gli altri ormoni, si vengono a manifestare specifici sintomi.
Ecco dunque i principali sintomi evocati dai PNET funzionali:
- Insulinoma: Triade di Whipple: sintomi ipoglicemici come offuscamento visivo e debolezza, glicemia inferiore a 50 mg/dL e risoluzione dei sintomi dopo assunzione di glucosio.
- Gastrinoma: ulcera peptica, dolore epigastrico, diarrea.
- VIPoma: diarrea acquosa, ipokaliemia, disidratazione ed acloridria (o acidosi).
- Glucagonoma: Intolleranza al glucosio, eruzione cutanea, stomatite, eritema migrante, cachessia.
- Somatostatinoma: Diabete mellito, colelitiasi, diarrea acquosa, steatorrea. [3]
Sintomi dei tumori neuroendocrini del pancreas di tipo non funzionale
Poiché i PNET di tipo non funzionale non sono interessati da una sindrome di iper-secrezione ormonale, essi possono essere riscontrati in maniera occasionale durante altre indagini cliniche; in alcuni casi, possono essere diagnosticati dopo un approfondimento di alcuni sintomi generici o sistemici, come:
- Diarrea, con frequenti evacuazioni acquose o poltacee nel corso della giornata;
- Ittero ostruttivo, visibile con colorazione giallastra della cute e della sclera degli occhi;
- Dispepsia;
- Senso di ripienezza post-prandiale;
- Percezione di un dolore sordo, gravativo o urente a livello addominale.
Diagnosi dei tumori neuroendocrini del pancreas
La diagnosi di un tumore neuroendocrino del pancreas può risultare ostica nella maggior parte dei casi, proprio perché i sintomi riportati dai pazienti sono sfumati e in genere riconducibili ad altre patologie più frequenti.
Quando si presentano degli evidenti disturbi a livello ormonale, sicuramente una visita endocrinologica approfondita può costituire un importante passo verso la diagnosi e la caratterizzazione clinica del PNET, da confermare con esami di laboratorio ed esami strumentali.
Pertanto, la diagnosi del tumore neuroendocrino del pancreas si può scomporre nelle seguenti fasi:
Visita medica ed esame obiettivo del paziente
Durante la visita medica, viene innanzitutto valutata l’anamnesi del paziente, con particolare riferimento all’anamnesi famigliare, al fine di poter individuare eventuali sindromi endocrine genetiche ricorrenti. Successivamente, durante l’esame obiettivo, il medico indaga circa il colorito della pelle del paziente, la dolorabilità evocata nei quadranti addominali e su ciascun sintomo riportato dal paziente stesso in fase anamnestica.
Esami di laboratorio generici
Sono di norma richiesti esami ematochimici di routine ed esami volti a saggiare la funzionalità pancreatica, come:
- Esame emocromocitometrico;
- Glicemia;
- Trigliceridemia;
- Indici aspecifici di flogosi, come PCR e VES;
- Amilasemia, lipasemia;
- Esame chimico-fisico e morfologico delle urine;
- Dosaggio dei grassi fecali.
Marcatori tumorali
In aggiunta a questi esami, viene poi richiesto il dosaggio dei markers tumorali, sia sierici che immunoistochimici, utili non solo nella fase di diagnosi ma anche nella successiva prognosi. I marcatori tumorali più ricercati sono:
- Cromogranina A;
- Enolasi neurone-specifica;
- Pancreastatina;
- Polipeptide pancreatico;
- Neurochinina A.
Tecniche di imaging
Le principali tecniche di diagnostica per immagini utilizzate sono:
- Ecografia endoscopica (EUS). Questa tecnica è utile soprattutto per rilevare lesioni di piccole dimensioni, da 2 millimetri in su e come guida per le procedure di biopsia.
- Tomografia computerizzata (TC) e Risonanza Magnetica Nucleare (RMN). Queste due tecniche sono molto utilizzate nell’individuazione dei PNET, vantano una buona sensibilità diagnostica e possono anche studiare la vascolarizzazione delle lesioni. Il limite è rappresentato dalle dimensioni della lesione: con un diametro inferiore ai 2 centimetri, la sensibilità diagnostica diminuisce sensibilmente.
- Scintigrafia del recettore della somatostatina (SRS), che sfrutta l’alta densità dei recettori della somatostatina da parte dei PNET.
- Tomografia a emissione di positroni (PET), dove si valuta l’attività delle cellule neoplastiche.
Biopsia del pancreas
L’agobiopsia del pancreas, spesso guidata dall’ecografia endoscopica, preleva un piccolo campione di tessuto pancreatico; questo campione di tessuto viene studiato tramite varie tecniche istochimiche, conferendo la certezza della presenza del tumore. [2],[3],[4]
Terapie e trattamenti dei tumori neuroendocrini del pancreas
La scelta della giusta terapia da impostare per intervenire direttamente sul tumore neuroendocrino del pancreas dipende dal tipo biologico del tumore nonché dalla sua capacità di evocare una sindrome ormonale iper-secernente.
Le terapie e i trattamenti più utilizzati sono:
- Interventi di chirurgia
La resezione chirurgica, spesso drastica, rappresenta la migliore soluzione per trattare con successo i tumori neuroendocrini del pancreas e, soprattutto, per prevenire la recidivizzazione. Oltre alla chirurgia radicale, può essere attuata anche la chirurgia citoriduttiva, soprattutto nei PNET di tipo funzionale.
- Chemioterapia citotossica
I tumori neuroendocrini del pancreas dimostrano una certa sensibilità agli agenti chemioterapici, soprattutto alla combinazione di platino ed etoposide. In genere i tumori più pericolosi, ossia i tumori non ben differenziati, sono meno resistenti alla chemioterapia citotossica rispetto ai tumori ben differenziati.
- Terapia ormonale con analoghi della somatostatina
La terapia ormonale può essere applicata con successo in pazienti affetti da tumori neuroendocrini del pancreas, mediante la somministrazione di sostanze, definite analoghe della somatostatina. Gli analoghi della somatostatina sono due molecole, l’octreotide e il lanreotide, si assumono per via iniettiva e, nei trial clinici, hanno dimostrato un buon controllo della proliferazione cellulare e della stabilizzazione metastatica.
- Radioterapia con recettori peptidici
Questa tecnica, attualmente approvata per l’uso in Europa, viene utilizzata nei tumori neuroendocrini del pancreas che tendono ad esprimere un’alta densità di recettori della somatostatina. [2],[3],[4]
Sopravvivenza relativa dei tumori neuroendocrini del pancreas
La sopravvivenza a 5 anni dal riscontro di tumore neuroendocrino viene determinata a seconda di quanto il cancro si sia diffuso e dalla presenza di eventuali metastasi, al momento della diagnosi.
Dunque,
- In caso di cancro localizzato, non esteso oltre i limiti anatomici del pancreas, la sopravvivenza relativa a 5 anni è del 93 %;
- In caso di cancro regionale, dove la neoplasia si estende oltre i confini del pancreas negli organi contigui, la sopravvivenza relativa a 5 anni è del 74 %;
- In caso di cancro disseminato, con presenza di metastasi in altri organi come polmoni, fegato e reni, la sopravvivenza relativa a 5 anni è del 24 %. [5]
Conclusioni
I tumori neuroendocrini del pancreas (PNET) rappresentano una classe assai eterogenea di tumori, più rari e allo stesso tempo meno aggressivi di altre neoplasie pancreatiche, come l’adenocarcinoma.
I PNET non funzionali sono più difficili da diagnosticare e, spesso, l’escissione per via chirurgica costituisce la strategia più efficace per garantire un buon controllo della patologia e prevenire il rischio di recidivizzazione; allo stato attuale, gli studi si stanno sempre più concentrando sull’eventuale impiego di nuove terapie, a base di analoghi della somatostatina, agenti neo-angiogenetici e inibitori di mTOR.

Fonti e note:
- [1] Anastasi G, Gaudio E, Tacchetti C. Trattato di Anatomia umana. Milano: Edi Ermes; 2019.
- [2] Sun J. Pancreatic neuroendocrine tumors. Intractable Rare Dis Res. 2017 Feb;6(1):21-28.
- [3] Ro C, Chai W, Yu VE, Yu R. Pancreatic neuroendocrine tumors: biology, diagnosis, and treatment. Chin J Cancer. 2013 Jun;32(6):312-24.
- [4] Vinik A, Perry RR, Casellini C, et al. Pathophysiology and Treatment of Pancreatic Neuroendocrine Tumors (PNETs). New Developments. 2018.
- [5] Cancer.org Survival Rates for Pancreatic Neuroendocrine Tumor. 2022.